Building the Materials Informatics System for Carbon Microbead Synthesis
- May 17
- 4 min read
Our AI/ML strategy starts with something unglamorous: years of manual R&D and manufacturing work.
Over the last 8+ years, we have built a dataset the hard way through real experiments, real production batches, and real operational decisions.
That's because we discover and manufacture tunable carbon-based microbeads using a continuous microfluidic droplet process — an approach that is inherently safer, more sustainable, and more cost-effective than conventional batch methods. In physical synthesis, there are no shortcuts. Every data point costs real time, real reagents, and real operator effort.
Now, we are building our materials informatics system to use data and AI/ML to understand not just that a
material works, but why it works, using that deep understanding to predict and accelerate what comes next.
Why does a continuous process matter for AI? In traditional materials science, scaling up from a lab flask

to a massive factory reactor introduces unpredictable physical changes - meaning an AI trained on lab data often struggles to generalize at a commercial scale. Because our continuous microfluidic architecture scales by using the exact same fluid dynamics throughout, our hardware is effectively software-controllable and scale-agnostic. This eliminates scaling artifacts, creating a consistent physical-digital loop - the right foundation for an AI-native discovery engine (Figure 1 shows what this looks like in practice — a researcher asking questions of the structured batch database in plain language, without writing a single line of code).

That foundation starts with data. We have now logged over 1,000 experimental and manufacturing batches across:
25+ distinct chemistry families and material compositions
Multiple steps in the manufacturing process
Multiple process variables at each stage - each affecting the final material
Structural, physical, chemical, and functional properties measured across every batch - from pore architecture and particle size, to chemical composition and electrical conductivity, to real-world separation and retention performance (Figure 2 shows a live view of this database — each row a real batch, each column a real measurement)
The data behind that foundation looks nothing like a software dataset. To put that in perspective, in software, datasets are often measured in millions or billions — generated
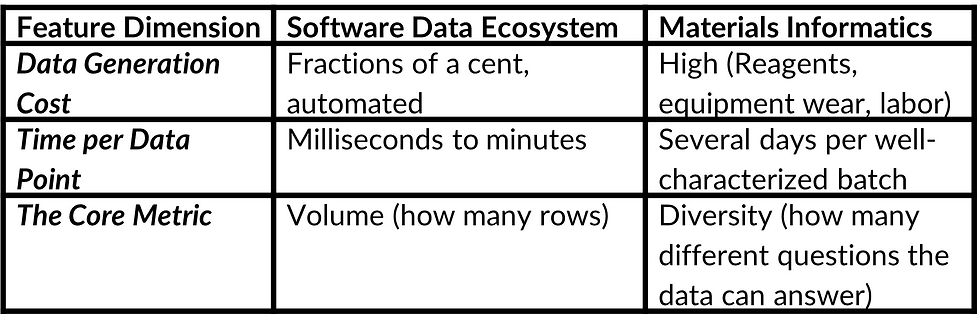
relatively quickly and at low cost. In physical materials synthesis, every data point requires real work: mixing chemicals, running a reactor, waiting for polymerization, washing, drying, sieving, and then characterizing the result with instruments that measure particle size, pore volume, and surface area. A single well-characterized batch can take several days and real money in reagents, equipment time, and operator effort (Figure 3 summarizes the key differences — cost per data point, time per experiment, and what the core metric actually measures).
So the right comparison is not to software datasets. It is to other physical science programs.
A typical academic paper in polymer or materials science draws conclusions from 10 to 30 experiments. A well-funded PhD thesis might document 50 to 150. The Cambridge Structural Database, arguably the world's most important materials dataset, took over 60 years of global scientific effort to reach one million crystal structure entries.
We built over 1,000 synthesis batches in 8 years, spanning 25 distinct chemistry families, across two
sites. That is not a small number. It is the result of deliberate, unglamorous, day-by-day experimental work. And it continues to grow with every batch we run.
There is one more thing that matters beyond the count. In materials science, the value of a dataset lies not in its size alone. It lies in diversity, context, and the range of questions it can answer. In machine learning on physical systems, diversity beats volume. A model trained on 1,000 experiments spanning 25 chemistry families, multiple particle size targets, multiple porosity architectures, and a wide range of process variables can generalize. Ten thousand repetitions of the same recipe on the same machine give you more data but less learning.

Our batches are not just production reruns. They are designed experiments that probe the edges of the process space across multiple sites, multiple years, different material systems, and a wide range of operating conditions. From droplet formation to final characterization, every stage adds complexity and generates data, making the science genuinely interesting. That depth gives us a strong foundation for AI/ML — because the platform is built on real complexity, not a simplified demo (Figure 4 shows the schema browser — 54 tables and growing, covering every stage from synthesis inputs to final performance characterization).
For more information on how our NanoPak-C All Carbon media or custom media services can address your pharmaceutical purification challenges or to request samples, please email us at inquiry@millennialscientific.com, call us at 855 388 2800, or fill in our online form.
References
Parente, M. J. and B. Sitharaman (2023). "Synthesis and Characterization of Carbon Microbeads." ACS omega 8(37): 34034-34043.
Greater Mastery Over Microbead Manufacture, Scientific American, 2024


